Module 3: Statistical Foundations for ExWAS
Conducting Exposome-Wide Association Studies
Overview
This module covers the statistical machinery behind ExWAS:
- Survey sampling concepts
svydesign()andsvyglm()vs.lm()- Multivariate regression
- Log transformation of skewed exposures
- Z-score standardization
- Multiple testing: Bonferroni vs. FDR
- Confounding and DAGs
- R-squared and delta R-squared
Why Special Statistics for NHANES?
NHANES uses complex survey sampling — not simple random sampling:
- Certain subpopulations are oversampled (e.g., minorities, elderly)
- Participants within the same PSU/stratum are correlated
- Each participant has a sampling weight reflecting how many people they represent
Consequence: Standard lm() produces biased standard errors and invalid p-values.
The Survey Package
The survey package in R handles complex survey designs:
Two key functions:
svydesign()— declare the survey designsvyglm()— fit regression models accounting for the design
Creating a Survey Design Object
- weights: how much each individual “counts” (inverse of selection probability)
- ids: clusters of correlated individuals (PSUs)
- strata: groups within which PSUs are sampled
svyglm() vs. lm(): Side by Side
Survey-weighted regression:
Ordinary regression (ignoring survey design):
Comparing Estimates
| method | term | estimate | std.error | p.value |
|---|---|---|---|---|
| svyglm | (Intercept) | 20.6623 | 0.1447 | 0 |
| svyglm | RIDAGEYR | 0.1455 | 0.0034 | 0 |
| lm | (Intercept) | 20.4280 | 0.0778 | 0 |
| lm | RIDAGEYR | 0.1416 | 0.0020 | 0 |
Note: coefficients may be similar, but standard errors differ — and that affects p-values.
When Does It Matter?
- For point estimates (coefficients), the difference is often modest
- For inference (p-values, confidence intervals), the difference can be substantial
- Always use survey-weighted methods when analyzing NHANES for valid inference
Multivariate Regression
Including multiple predictors in a single model:
\[\text{BMI} = \alpha + \beta_1 \cdot \text{Age} + \beta_2 \cdot \text{Sex} + \beta_3 \cdot \text{Race} + \beta_4 \cdot \text{Income} + \epsilon\]
Multivariate Results
| term | estimate | std.error | p.value |
|---|---|---|---|
| (Intercept) | 20.4261 | 0.2579 | 0.0000 |
| RIDAGEYR | 0.1524 | 0.0034 | 0.0000 |
| male | -0.2090 | 0.1329 | 0.1302 |
| black | 1.5605 | 0.1810 | 0.0000 |
| mexican | 0.9928 | 0.1902 | 0.0000 |
| other_hispanic | 0.5532 | 0.2813 | 0.0620 |
| other_eth | -0.0129 | 0.6124 | 0.9833 |
| INDFMPIR | -0.0620 | 0.0569 | 0.2879 |
Interpreting Multivariate Coefficients
Each coefficient represents the association holding all other variables constant:
RIDAGEYR: change in BMI per 1-year increase in age, adjusted for sex, race, incomemale: difference in BMI for males vs. females, adjusted for age, race, incomeblack,mexican, etc.: difference in BMI relative to white (reference group)
Exploring an Exposure: Blood Lead
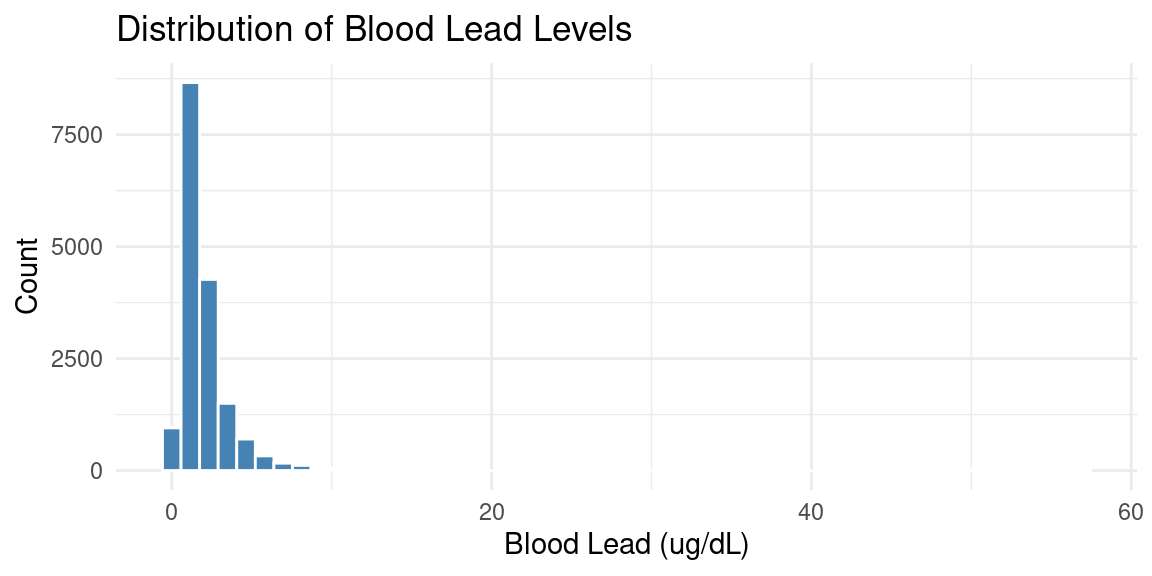
The distribution is right-skewed — most values are low with a long right tail.
Why Log-Transform?
- Many exposure biomarkers have right-skewed distributions
- Log transformation makes the distribution more symmetric
- Improves linear model assumptions (normality of residuals)
- Common practice in environmental epidemiology
Log-Transforming Lead
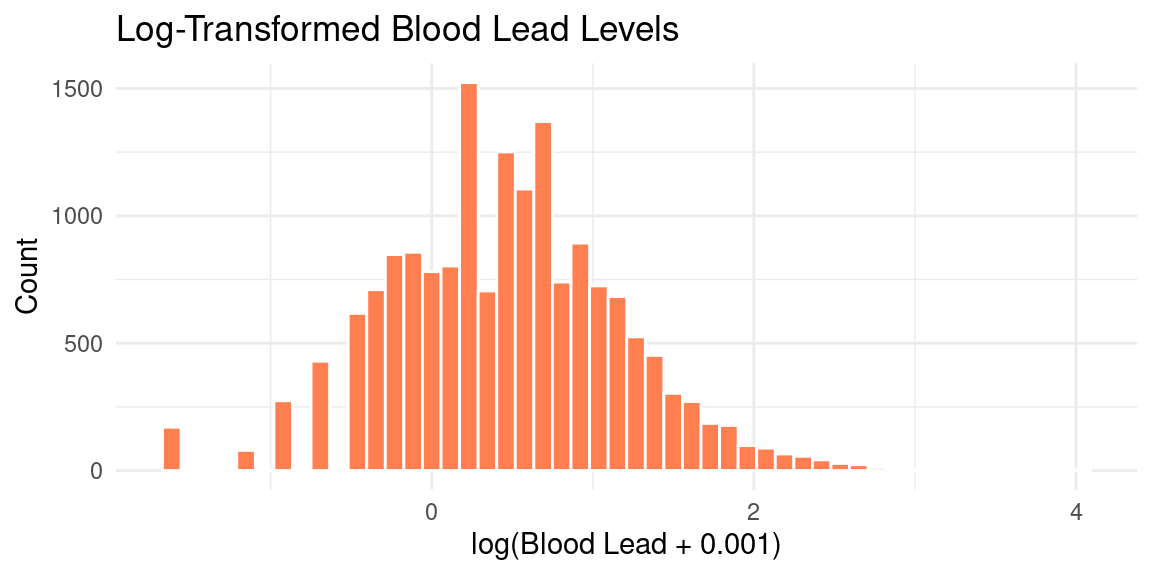
Why Add 0.001?
- Some exposure values may be zero (below detection limit)
- \(\log(0) = -\infty\), so we add a small constant
log(LBXBPB + 0.001)ensures all values are defined- The constant (0.001) is small enough to not affect non-zero values substantially
Interpreting Log-Transformed Coefficients
| term | estimate | std.error | statistic | p.value |
|---|---|---|---|---|
| (Intercept) | 98.294 | 0.628 | 156.535 | 0 |
| log(LBXBPB + 0.001) | 4.322 | 0.664 | 6.511 | 0 |
A 1-unit change in log(exposure) corresponds to a ~2.7-fold change in the raw exposure (since \(e^1 \approx 2.72\)).
Z-Score Standardization (Scaling)
Different exposures have different units (ug/dL, ng/mL, etc.). To compare across exposures:
\[z = \frac{x - \bar{x}}{s_x}\]
NHData.train <- NHData.train %>%
mutate(lead_scaled = scale(log(LBXBPB + 0.001)))
NHData.train %>%
summarize(
raw_mean = mean(log(LBXBPB + 0.001), na.rm = TRUE),
raw_sd = sd(log(LBXBPB + 0.001), na.rm = TRUE),
scaled_mean = mean(lead_scaled, na.rm = TRUE),
scaled_sd = sd(lead_scaled, na.rm = TRUE)
) %>%
kable(digits = 3) %>% kable_styling()| raw_mean | raw_sd | scaled_mean | scaled_sd |
|---|---|---|---|
| 0.46 | 0.713 | 0 | 1 |
Why Standardize?
After scaling:
- Mean = 0, SD = 1
- Coefficients represent the change in outcome per 1 SD change in the exposure
- All exposure associations are on the same scale
- Essential for comparing effect sizes across different exposures in ExWAS
The ExWAS Model
For each exposure \(E_j\), we fit:
\[\text{Phenotype} = \alpha + \beta_j \cdot \text{scale}(\log(E_j + 0.001)) + \gamma_1 \cdot \text{Age} + \gamma_2 \cdot \text{Sex} + \gamma_3 \cdot \text{Race} + \gamma_4 \cdot \text{Income} + \epsilon\]
- \(\beta_j\): effect of a 1 SD change in log-exposure on the phenotype
- Adjusted for confounders (age, sex, race, income)
Multiple Testing Problem
When testing \(m\) hypotheses simultaneously:
- At \(\alpha = 0.05\), we expect \(0.05 \times m\) false positives by chance
- With \(m = 160\) exposures: ~8 false positives expected
- We need multiple testing correction
Bonferroni Correction
The simplest approach — divide \(\alpha\) by the number of tests:
\[\alpha_{\text{Bonf}} = \frac{\alpha}{m} = \frac{0.05}{160} = 0.000313\]
- Controls the Family-Wise Error Rate (FWER)
- Probability of any false positive \(\leq \alpha\)
- Very conservative — may miss true associations
Benjamini-Hochberg FDR
A less conservative approach controlling the False Discovery Rate:
- Rank p-values from smallest to largest: \(p_{(1)} \leq p_{(2)} \leq \cdots \leq p_{(m)}\)
- Find the largest \(k\) such that \(p_{(k)} \leq \frac{k}{m} \cdot \alpha\)
- Reject all hypotheses \(H_{(1)}, \ldots, H_{(k)}\)
- Controls the expected proportion of false discoveries among rejected hypotheses
- Less conservative than Bonferroni
- Preferred for ExWAS
FDR in R
# Simulated p-values for demonstration
set.seed(42)
pvalues <- c(runif(10, 0, 0.001), runif(150, 0, 1)) # 10 "real" + 150 null
# Bonferroni
bonf_adjusted <- p.adjust(pvalues, method = "bonferroni")
# Benjamini-Hochberg FDR
fdr_adjusted <- p.adjust(pvalues, method = "BH")
tibble(
raw_p = sort(pvalues)[1:5],
bonferroni = sort(bonf_adjusted)[1:5],
fdr = sort(fdr_adjusted)[1:5]
) %>% kable(digits = 6) %>% kable_styling()| raw_p | bonferroni | fdr |
|---|---|---|
| 0.000135 | 0.021547 | 0.01363 |
| 0.000239 | 0.038223 | 0.01363 |
| 0.000286 | 0.045782 | 0.01363 |
| 0.000519 | 0.083055 | 0.01363 |
| 0.000642 | 0.102679 | 0.01363 |
Bonferroni vs. FDR: Comparison
tibble(raw = sort(pvalues), bonf = sort(bonf_adjusted), fdr = sort(fdr_adjusted),
rank = 1:length(pvalues)) %>%
pivot_longer(cols = c(bonf, fdr), names_to = "method", values_to = "adjusted_p") %>%
ggplot(aes(x = rank, y = adjusted_p, color = method)) +
geom_point(alpha = 0.5) +
geom_hline(yintercept = 0.05, linetype = "dashed") +
scale_y_log10() +
labs(x = "Rank", y = "Adjusted p-value (log scale)", title = "Bonferroni vs. FDR") +
theme_minimal()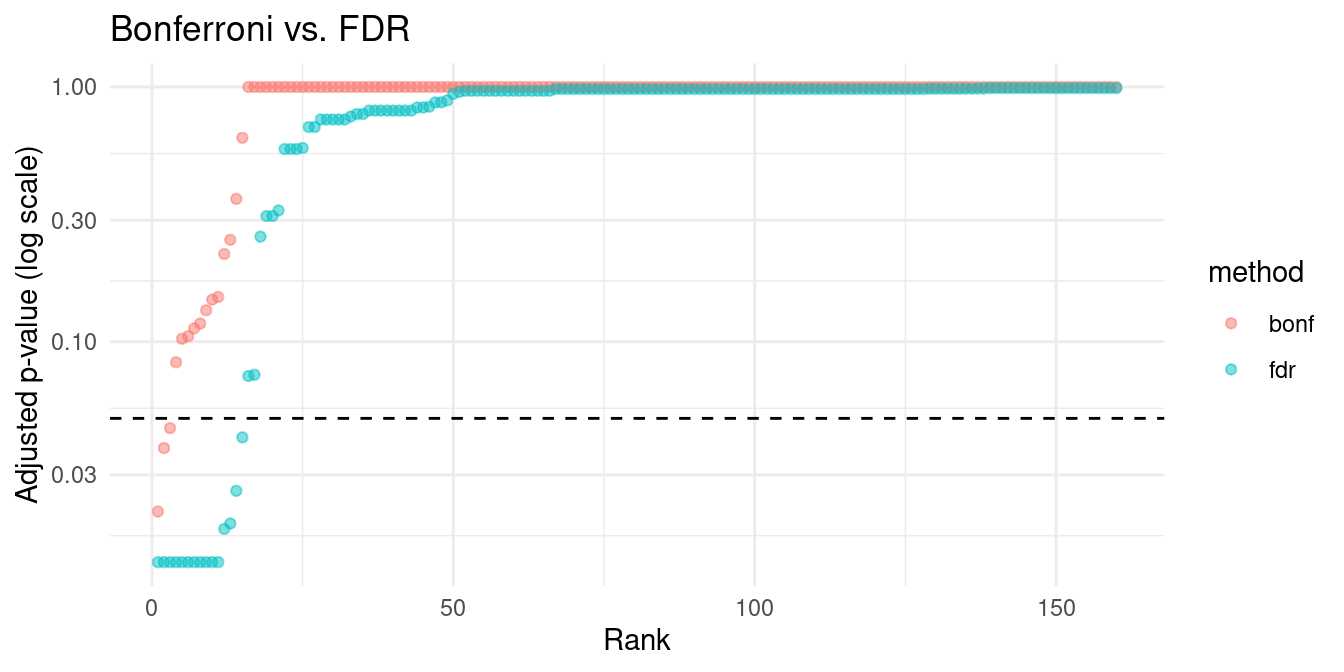
Confounding
A confounder C is a variable that:
- Is associated with the exposure (E)
- Is associated with the outcome (P)
- Is NOT on the causal path from E to P
If we don’t adjust for C, the E-P association is biased.
Directed Acyclic Graphs (DAGs)
DAGs represent causal relationships:
C
/ \
v v
E P- C → E: confounder causes exposure level
- C → P: confounder causes phenotype
- Without adjusting for C, we see a spurious E-P association
Example: Age (C) affects both lead exposure (E) and BMI (P).
Adjusting for Confounders
In our ExWAS model, we adjust for:
| Confounder | Why? |
|---|---|
| Age | Affects both exposure levels and health outcomes |
| Sex | Biological differences in metabolism and exposure |
| Race/Ethnicity | Sociostructural differences in exposure and health |
| Income (INDFMPIR) | Socioeconomic factors affect exposure and health |
The Confounding Problem in ExWAS
A fundamental limitation of screening approaches like ExWAS:
Each exposure-phenotype pair has a different confounding structure — but we apply the same covariate set to all tests.
Lead → BMI: confounders = age, SES, housing, occupation
Vitamin D → BMI: confounders = age, SES, physical activity, sun exposure
Cotinine → BMI: confounders = age, SES, alcohol use, diet qualityThere is no single DAG for the exposome — there are hundreds of DAGs, one per association.
Why This Matters
Using a “one-size-fits-all” covariate set means:
- Under-adjustment: missing a key confounder for some associations → false positives
- Over-adjustment: conditioning on a collider or descendant of the exposure → bias
- Heterogeneous bias: the direction and magnitude of bias differs across exposures
Example: Adjusting for BMI when testing diet → glucose may block a causal path (over-adjust), while failing to adjust for physical activity when testing vitamin D → BMI may leave confounding open (under-adjust).
Implications for ExWAS
This does not invalidate ExWAS — but it means:
- ExWAS results are hypothesis-generating, not causal claims
- The same covariate set is a practical necessity, not an ideal
- Sensitivity analysis across multiple adjustment models (Module 4) helps assess robustness
- Top hits should be followed up with exposure-specific DAGs and targeted analyses
- Advanced methods can help mitigate this bias (Module 7)
R-Squared (\(R^2\))
\(R^2\) measures the proportion of variance in the outcome explained by the model:
\[R^2 = 1 - \frac{SS_{\text{res}}}{SS_{\text{tot}}}\]
Delta R-Squared (\(\Delta R^2\))
How much additional variance does the exposure explain beyond confounders?
\[\Delta R^2 = R^2_{\text{full}} - R^2_{\text{base}}\]
# Base model (confounders only)
r2_base <- summary(lm(BMXBMI ~ RIDAGEYR + male +
black + mexican + other_hispanic + other_eth +
INDFMPIR,
data = NHData.train))$r.squared
# Full model (confounders + exposure)
r2_full <- summary(lm(BMXBMI ~ scale(log(LBXBPB + 0.001)) +
RIDAGEYR + male +
black + mexican + other_hispanic + other_eth +
INDFMPIR,
data = NHData.train))$r.squared
tibble(r2_base = r2_base, r2_full = r2_full,
delta_r2 = r2_full - r2_base) %>%
kable(digits = 5) %>% kable_styling()| r2_base | r2_full | delta_r2 |
|---|---|---|
| 0.23431 | 0.25109 | 0.01678 |
Putting It Together: The ExWAS Pipeline
- Set up survey design with
svydesign() - For each exposure: log-transform and standardize
- Fit
svyglm()adjusting for confounders - Collect estimates, standard errors, p-values
- Correct p-values with Benjamini-Hochberg FDR
- Compare \(\Delta R^2\) across exposures
- Replicate in independent dataset
Summary
- Survey-weighted methods (
svydesign,svyglm) are essential for NHANES - Log-transform skewed exposure distributions
- Standardize (Z-score) to compare effect sizes across exposures
- Multiple testing correction: FDR preferred over Bonferroni for ExWAS
- Confounders must be identified and adjusted for (use DAGs)
- Confounders differ per association — a major limitation of one-size-fits-all screening
- \(\Delta R^2\) quantifies the unique contribution of each exposure
What’s Next?
Module 4: The nhanespewas Package
- Installing and connecting to the nhanespewas database
- Exploring exposure and phenotype catalogs
- Understanding adjustment models
- First ExWAS with
pe_flex_adjust()
Supported By
This course is supported by the National Institutes of Health (NIH):
- National Institute of Environmental Health Sciences (NIEHS): R01ES032470, U24ES036819
- National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK): R01DK137993